
MDock
Overview
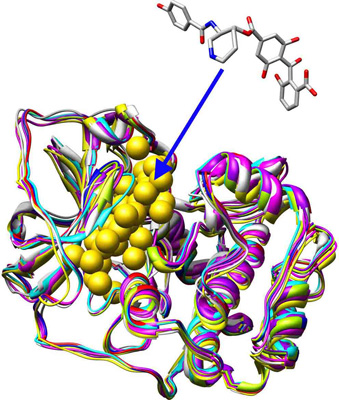 MDock is an automated molecular docking software which can simultaneously docking ligands against multiple protein structures/conformations by using the ensemble docking algorithm (Huang and Zou, 2007a,b). It supports docking/optimizing ligand(s) against either a single protein structure or an ensemble of multiple protein structures and score calculations for given protein-ligand complexes. The energy function used in MDock is the knowledge-based scoring function, ITScore (Huang and Zou, 2006a,b).
MDock is an automated molecular docking software which can simultaneously docking ligands against multiple protein structures/conformations by using the ensemble docking algorithm (Huang and Zou, 2007a,b). It supports docking/optimizing ligand(s) against either a single protein structure or an ensemble of multiple protein structures and score calculations for given protein-ligand complexes. The energy function used in MDock is the knowledge-based scoring function, ITScore (Huang and Zou, 2006a,b).
MDock uses the sphere-ligand matching algorithm of UCSF DOCK to generate possible ligand conformations. Therefore, a UCSF DOCK license is required to generate the sphere points that represent a negative image of the protein to be matched with ligand atomic centers, for the use of MDock. UCSF DOCK is free for academic users. Files prepared for UCSF DOCK 4.0 can be applied to MDock without modification. In general, docking preparation would be easier for MDock than for UCSF DOCK, because MDock does not require adding hydrogens and charges for proteins and ligands.
In parallel to the method development for ligand-protein docking, studies on protein-protein docking are also under way (Huang and Zou, 2008).
Availability
The MDock package is available free of charge for academic users. To obtain an academic user license, please download the MDock academic license file, fill out, and fax back to us. Upon approval, an FTP site with a temporary password for downloading MDock will be sent to you.
For commercial or other non-academic users, please send emails to mu-dcrc-mdock-license@missouri.edu for details of access conditions.
References
Huang SY, Zou XQ.
An iterative knowledge-based scoring function to predict protein-ligand interactions: I. Derivation of interaction potentials.
Journal of Computational Chemistry,27:1866-1875, 2006a.
Huang SY, Zou XQ.
An iterative knowledge-based scoring function to predict protein-ligand interactions: II. Validation of the scoring function.
Journal of Computational Chemistry , 27:1876-1882, 2006b.
Huang SY, Zou XQ.
Ensemble docking of multiple protein structures: Considering protein structural variations in molecular docking.
Proteins, 66:399-421, 2007a.
Huang SY, Zou XQ.
Efficient molecular docking of NMR structures: Application to HIV-1 protease.
Protein Science, 16:43-51, 2007b.
Huang SY, Zou XQ.
An iterative knowledge-based scoring function for protein-protein recognition.
Proteins, 72:557-579, 2008.
Huang SY, Zou XQ.
A non-redundant structure dataset for benchmarking protein-RNA computational docking.
Journal of Computational Chemistry, doi: 10.1002/jcc.23149.